
| 発表のポイント |
|---|
|
発表概要
- 細胞増殖の厳密な制御は人体の正常な発生のみならず、細胞の癌化を防ぐ上でも極めて重要です。ヒトの細胞増殖はERK経路(注1)と呼ばれる3つの蛋白質リン酸化酵素(Raf−MEK−ERK)からなる情報伝達システムによって制御されていますが、実際に様々な癌でMEK1を始めとするERK経路の構成因子に遺伝子変異が認められ、この経路の異常な活性化を招いて発癌の原因となることが知られています。また近年、発生異常を特徴とする遺伝性疾患「先天性Ras/MAPK症候群(RASopathy)(注2)」においても、MEK1遺伝子の変異が見出され、MEK1の異常が、癌のみならず発生異常の原因となることも明らかとなりました。しかし、MEK1という同一の分子に生じる変異が、どの様にして癌と発生異常という異なる臨床像をもつ別個の疾患を誘発するのか、そのメカニズムは全く分かっていませんでした。
今回、東京大学医科学研究所分子シグナル制御分野の武川睦寛教授、久保田裕二助教らの研究グループは、癌やRASopathyで見出されるMEK1変異体の詳細な解析を行い、これら2疾患で認められる各MEK1変異体は、原疾患に依存してその活性化機構、酵素活性の強度、および生物学的性質が大きく異なっていることを発見しました。即ち、癌由来のMEK1変異体は、異常な自己リン酸化能(自分自身をリン酸化して活性化する能力)を獲得しており、極めて強い酵素活性と細胞癌化能を有するのに対し、RASopathy由来の変異体は自己リン酸化能を有しておらず、中程度の酵素活性を示し、発癌能にも乏しいことを見出しました。さらにこの様なMEK1変異体の疾患特異的な性質の違いが、細胞内の情報伝達と遺伝子発現プロファイルに異なるタイプの異常を引き起こして、癌と発生異常という別個の臨床像が導かれていることを明らかにしました。
加えて研究グループは、この解析の過程で、PHLDA1およびPHLDA2という分子が、癌特異的に高発現しており、このことが分子標的抗癌剤に対する癌細胞の治療抵抗性獲得に寄与していることを見出しました。また実際に、PHLDA1/2の発現を人工的に制御することで、分子標的抗癌剤の抗腫瘍効果が飛躍的に高まることを動物実験で確認しました。
本研究成果は、癌およびRASopathyの病態解明とその克服に向けて重要な手がかりを与える知見であり、今後これらの成果を活用することで、癌やRASopathyに対する早期診断法や、より効果的な治療法の開発が期待されます。
本成果は2022年7月13日(英国時間)、英国科学雑誌「Nature Communications」に掲載されました。なお本研究は、北海道大学の野田展生教授、東京大学の中井謙太教授、松原大祐先生(現筑波大学教授)、微生物化学研究所の百瀬功主席研究員らとの共同研究として行われました。
発表内容
細胞増殖の厳密な制御は、人体の正常な発生のみならず、発癌を抑制する上でも極めて重要です。ヒト細胞の増殖は、ERK経路という3つの蛋白質リン酸化酵素(Raf−MEK−ERK)からなるシグナル伝達経路によって制御されており、またその破綻は発癌の原因となります。実際に、この経路の上流に位置する増殖因子受容体(EGFR等)、Ras、Rafなどの分子は、様々な癌で遺伝子変異が認められる癌遺伝子であり、ERK経路を異常に活性化して無秩序な細胞増殖を惹起し、発癌を導きます。また、これら癌遺伝子の働きを抑制してERK経路を阻害する薬剤は、分子標的抗癌剤として、広く癌治療に用いられています。
近年、癌臨床サンプルを用いた解析から、ERKの直接的活性化因子であるMEK1の遺伝子変異が多数の癌で見出され、MEK1も癌遺伝子として機能し、その異常が発癌の原因となることが報告されました(図1a)。さらに最近、発生異常(心筋症、精神遅滞など)を特徴とする遺伝性疾患「先天性Ras/MAPK症候群(RASopathy)」においても、MEK1遺伝子の変異が多数同定され、MEK1の遺伝子変異が、癌のみならず発生異常の原因となることも明らかになりました。しかし、MEK1という同一の分子に生じる変異が、どの様にして癌と発生異常という全く異なる臨床像をもつ別個の疾患を誘発するのか、そのメカニズムは分かっていませんでした。
今回、東京大学医科学研究所の武川睦寛教授・久保田裕二助教らの研究グループは、癌やRASopathyで見出されるMEK1変異体の詳細な解析を行い、これら2疾患で認められるMEK1変異が、異なる病態を導くメカニズムを明らかにしました。また、この解析の過程で、癌細胞が分子標的抗癌剤に対して抵抗性を獲得する新たな機構を発見するとともに、この機構を人工的に遮断することで、抗癌剤の治療効果が飛躍的に高まることを動物実験で確認しました。
まず研究チームは、癌とRASopathyの両疾患で、MEK1遺伝子変異の部位やアミノ酸変化の種類が異なることに着目し、各MEK1変異体の性質に何らかの違いがあるか検討を行いました。MEK1は通常、増殖因子等の刺激に応じて、上流のRafによってリン酸化されると活性化することが知られています。しかし、疾患由来のMEK1変異体は、その全てが無刺激の状態でも高い酵素活性を示す「恒常的活性化型」となっていることが分かりました。さらに興味深いことに、各変異体の活性強度や性質は、原疾患に依存して大きく異なっていることを見出しました(図1b)。即ち、癌由来のMEK1変異体は、異常な自己リン酸化能(自分自身をリン酸化して活性化する能力)を獲得しており、極めて強い酵素活性と細胞癌化能を有するのに対し、RASopathy由来の変異体は自己リン酸化能を有しておらず、リン酸化非依存的に活性化して中程度の酵素活性を示し、発癌能にも乏しいことを発見しました。
-
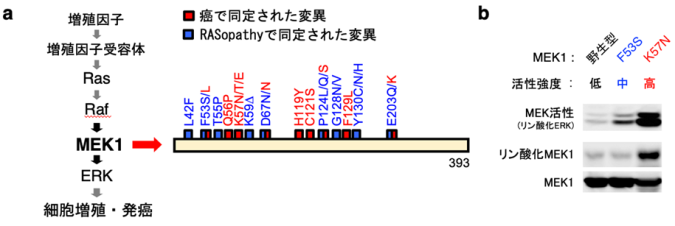
図1: 癌およびRASopathyで認められるMEK1変異体は異なる性質を示す
a.(左)ERK経路の模式図。(右)癌およびRASopathyにおけるMEK1変異。
b.MEK1変異体の酵素活性とリン酸化状態。癌由来MEK1変異体(K57N)は、異常な自己リン酸化能を獲得して、強い酵素活性と細胞癌化能を有するのに対し、RASopathy由来変異体(F53S)は自己リン酸化能を有しておらず、中程度の酵素活性を示し、発癌能にも乏しい。次に、MEK1変異体の異常な活性化機構を解明するため、変異型MEK1蛋白質を精製して結晶構造解析を実施し、正常なMEK1分子の構造と比較しました(図2)。
その結果、変異型MEK1においては、1)活性化ループと呼ばれる領域が、分子の外側に向かって大きく開いており、2)これによって、野生型MEK1では活性化ループで隠されていた「基質結合領域」や「酵素活性中心」が解放されて、基質(ERK)をリン酸化し易い状態になっていることが分かりました。加えて、3)野生型MEK1で認められる、近接した3つのアミノ酸残基(K57/H119/F129)間の相互作用(水素結合)が、癌由来の変異体では完全に消失しており、このことが異常な自己リン酸化能と発癌能を獲得する主因であることを発見しました。また、実際に様々なヒト癌において、これら3つのアミノ酸残基のいずれかに点変異が認められることを確認しました。
-
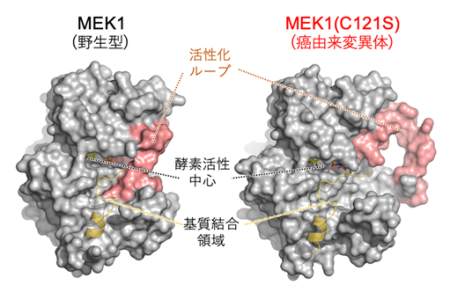
図2:野生型および癌由来MEK1変異体の立体構造
野生型と比べて、癌由来変異体では、活性化ループ(赤色部分)が開いており、酵素活性中心と基質(ERK)結合領域が露出して、酵素反応が起こり易い活性化状態となっている。次に、これら性質の異なる2種類のMEK1変異体が、細胞内でERK経路の制御と遺伝子発現にどの様な影響を与えるのか解析を行いました。その結果、RASopathy由来の変異体を発現する細胞は、野生型細胞と比べて、無刺激時におけるERK活性の亢進は軽度であるものの、増殖因子刺激後に起こるERKの活性化と、それに伴うERKの細胞質から核への移行が有意に増強し、その持続時間も延長することが分かりました(図3)。 - またそれに伴って、細胞内の遺伝子発現パターンが変化し、心筋症や精神遅滞などRASopathyの臨床症状に関連する遺伝子群の発現が選択的に亢進することが分かりました。一方、癌由来の変異体を発現する細胞では、無刺激の状態でもERKが強く活性化して核内に集積しており、その結果、特定の転写因子が常に活性化された状態となって、発癌および癌の進展に関わる遺伝子が多数発現誘導されることを見出しました。
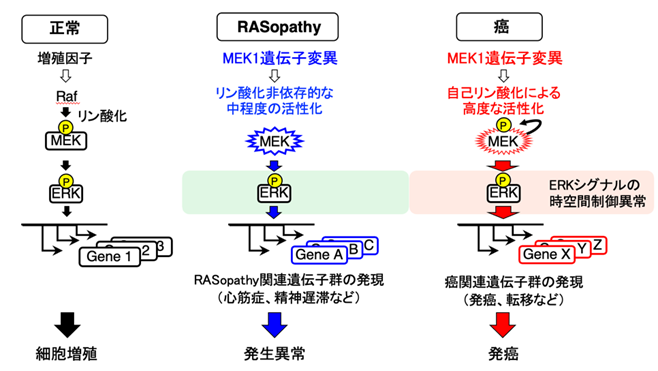
図3:MEK1遺伝子の変異がRASopathyや癌を誘発するメカニズム
癌およびRASopathy由来のMEK1変異体は、活性化機構や性質が原疾患に依存して異なっている。その結果、細胞内の情報伝達および遺伝子発現プロファイルに異なるタイプの異常を引き起こし、癌と発生異常という別個の疾患が導かれる。
- これらの結果から、癌およびRASopathyで認められるMEK1変異体の性質の違いが、ERKシグナルの時空間制御と、細胞内の遺伝子発現プロファイルに異なるタイプの異常を引き起こし、発癌と発生異常という別個の疾患を導いていることが明らかとなりました。
さらに研究グループは、上述の解析過程で、RASopathy由来のMEK1変異体(中程度のERK活性)では発現量が変化せず、癌由来のMEK1変異体(強力かつ恒常的なERK活性)を持つ細胞においてのみ、発現誘導される遺伝子が一定数存在することを見出しました。この様な遺伝子は癌細胞特異的に発現しており、癌の病態に深く関与する可能性が想定されます。そこで、これらの遺伝子の中から、特にPHLDA1およびPHLDA2という分子に着目して詳細な解析を行いました。まず、実際のヒト癌組織におけるPHLDA1/2の発現を検証したところ、各種癌遺伝子(EGFR、Ras、Raf、MEK1等)に変異を有し、ERK活性の高い様々な癌(肺・大腸・膵癌、悪性黒色腫など)で、PHLDA1/2が共に高発現していることを確認しました。- さらに、これらの分子の機能について解析を進めた結果、癌細胞におけるPHLDA1/2の高発現が、発癌過程と癌治療において「諸刃の剣」として機能していることを見出しました(図4a)。
- 即ち、1)PHLDA1/2は、元来、細胞の生存シグナル(AKT経路:注3)を阻害する作用を持つ分子であり、ERK活性の高い癌で高発現することで、癌細胞の生存を抑制し、腫瘍の増大を一定程度抑えているが、その一方で、2)癌治療の際に、分子標的抗癌剤(ERK経路阻害剤)を患者に投与すると、癌細胞内のERK活性が低下してPHLDA1/2の発現が急速に消失してしまうため、これらの分子による抑制が解除されてAKT(生存)経路が強く活性化し、癌細胞が細胞死を回避する様になって抗癌剤の効果が減弱してしまうことが分かりました。実際に、癌細胞内にPHLDA1/2を強制的に導入してその発現を維持してやると、分子標的抗癌剤の抗腫瘍効果が著しく高まることを動物実験で確認しました(図4b)。
- 上記結果から、PHLDA1/2の発現を人工的に制御することが出来れば、分子標的抗癌剤の効果を飛躍的に高めることが可能であることが分かりました。そこで最後に、PHLDA1/2の発現を、ERK活性非依存的に亢進させる薬剤が存在するか、探索を行いました。その結果、既存薬ボルテゾミブ(注4)が、転写および転写後調節の両レベルで、PHLDA1/2の発現を増強する作用をもつことを発見しました。また実際に、分子標的抗癌剤(ERK経路阻害剤)とボルテゾミブを、癌細胞に同時に投与すると、ERK活性が低下してもPHLDA1/2の発現が維持されて生存(AKT)シグナルが抑制されたままとなり、抗癌作用(癌細胞の細胞死)が有意に高まることを確認しました(図4c)。
-
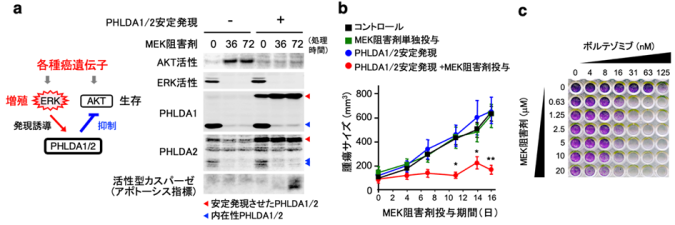
図4:分子標的抗癌剤によるPHLDA1/2の発現低下が、癌細胞の抗癌剤抵抗性獲得に関与する
a. (左)PHLDA1/2を介したERK-AKT経路の癌特異的クロストーク。(右)レーン1-3:MEK阻害剤(分子標的抗癌剤トラメチニブ)の投与により、癌細胞内のERK活性が低下すると、PHLDA1/2の発現が消失してAKT(生存)経路が活性化してしまう。このため癌細胞は死(アポトーシス)を免れて抗癌剤に抵抗性を示す。レーン4-6:一方、癌細胞にPHLDA1/2を導入して人工的に安定発現させると、ERK活性が低下してもAKTが抑制されたままとなり、癌細胞にアポトーシスが誘導される 。
b. 担癌マウスを用いた癌治療実験。PHLDA1/2を安定発現させた癌を持つマウスに、MEK阻害剤を経口投与すると、腫瘍の増大が著しく抑制された(赤丸)。
c. 癌細胞(紫色)にMEK阻害剤とボルテゾミブを同時投与すると、ERK活性が低下してもPHLDA1/2の発現が維持され、MEK阻害剤の抗癌作用(細胞死誘導)が著しく増強する。
- 本研究により、癌およびRASopathyで見出されるMEK1変異体は、原疾患に依存して異なる活性化機構と性質を有しており、その結果、細胞内の遺伝子発現パターンに大きな違いが生じて、発癌と発生異常という別個の疾患が導かれていることが明らかとなりました。また、網羅的遺伝子解析から、癌およびRASopathy特異的に発現する多数の分子を同定することに成功しました。これらの中には、細胞外に分泌される分子も多く含まれており、癌に対する新たな血清診断マーカーや、RASopathyに対する出生前羊水診断マーカーになり得ると考えられます。さらに今回、様々な癌(肺・大腸・膵臓癌、悪性黒色腫など)においてPHLDA1/2が高発現していること、また、分子標的抗癌剤の投与に伴うその発現低下が、癌細胞の抗癌剤抵抗性を導く主因の一つであることを見出しました。今後、本研究の知見を活用することで、癌やRASopathyに対する早期診断マーカーや、より効果的な癌治療法の開発が期待されます。
発表雑誌
雑誌名:「Nature Communications」(7月13日 オンライン版)論文タイトル:Qualitative differences in disease-associated MEK mutants reveal molecular signatures and aberrant signaling-crosstalk in cancer
著者: Yuji Kubota, Yuko Fujioka, Ashwini Patil, Yusuke Takagi, Daisuke Matsubara, Masatomi Iijima, Isao Momose, Ryosuke Naka, Kenta Nakai, Nobuo Noda, and *Mutsuhiro Takekawa(*責任著者)
DOI番号:10.1038/s41467-022-31690-w
アブストラクトURL:https://www.nature.com/articles/s41467-022-31690-w
-
問い合わせ先
東京大学医科学研究所 分子シグナル制御分野
教授 武川睦寛(たけかわ むつひろ)
https://www.ims.u-tokyo.ac.jp/imsut/jp/lab/basicmedicalsciences/section02.html
<報道に関するお問い合わせ>
東京大学医科学研究所 国際学術連携室(広報)
https://www.ims.u-tokyo.ac.jp/imsut/jp/
-
用語解説
3種類の蛋白質リン酸化酵素(Raf-MEK-ERK)により構成されるシグナル伝達モジュール。増殖因子に応答して、上流から下流への連続的なリン酸化反応を経て活性化され、細胞増殖を制御する。MEKやRaf、さらにその上流のRas、増殖因子受容体はいずれも癌遺伝子であり、ERKを異常に活性化して発癌を招く。
(注2) 先天性Ras/MAPK症候群(RASopathy):
常染色体優性遺伝性疾患であるCFC(cardio-facio-cutaneous)症候群、ヌーナン症候群、コステロ症候群、LEOPARD症候群の総称。ERK経路を制御する様々な遺伝子の変異によって発症し、心疾患(心筋症)、精神遅滞、骨格異常、特異的顔貌、皮膚症状などの症状を呈する。
(注3) AKT経路:
増殖因子やインスリン等によって活性化されるシグナル伝達経路であり、細胞の死(アポトーシス)を強く阻害して、生存を促進する。AKT経路の活性化は癌細胞の生存を促し、癌の更なる悪性化や抗癌剤抵抗性に関与する。
(注4) ボルテゾミブ:
タンパク質分解酵素複合体(プロテアソーム)の特異的阻害剤。多発性骨髄腫などの治療薬として用いられている。