
新規ERK基質分子MCRIP1による上皮間葉転換の制御~ERKシグナルによる癌抑制遺伝子のジーン・サイレンシング~
新規ERK基質分子MCRIP1による上皮間葉転換の制御~ERKシグナルによる癌抑制遺伝子のジーン・サイレンシング~
上皮間葉転換(EMT)は、様々な増殖因子刺激(TGFβ、EGF、HGFなど)に応じて、上皮細胞が上皮マーカーであるE-カドヘリン(細胞同士を連結して繋ぎ止める分子)の発現を失い、運動能の高い間葉系細胞の性質を獲得する現象です。EMTは器官形成や創傷治癒、組織線維化など、様々な生理的・病理的生命現象に関与しますが、癌細胞が高い運動能を獲得して浸潤・転移する際にも重要であることが知られています。従って、EMTの制御機構、特にEMTの最も重要なステップであるE-カドヘリンの発現抑制機構を解明することは、癌悪性化メカニズムを理解し、新たな治療法を開発する上でも極めて重要です。しかしながら、その分子機構には不明な点が多く残されています。
癌細胞で異常なEMTが誘発される一因として、これまでにERK経路(Raf-MEK-ERK)の活性化が重要であることが報告されています。実際に、この経路の上流に位置するRasやRafなどの分子は、様々な癌において高率に遺伝子変異が見出される癌遺伝子であり、ERK経路を異常に活性化して細胞を癌化させると共に、E-カドヘリンの発現をも抑制してEMTを誘導することが明らかにされています。その一方で、E-カドヘリンの発現抑制に、転写抑制共役因子であるCtBPが関与することも示されています。CtBPは、核内で様々なヒストン修飾酵素[脱アセチル化酵素(HDAC)やメチル化酵素(HMT)等]と複合体を形成する分子ですが、自分自身ではDNAと結合する能力を持たないため、普段は核質内に局在しています。しかし、細胞が増殖因子などで刺激されると、CtBPは染色体DNA上に存在する転写抑制因子(ZEBなど)と結合して標的遺伝子のプロモーター領域にリクルートされ、周辺のヒストン修飾を変化させてE-カドヘリンを始めとする様々な遺伝子の転写を抑制することが知られています。従って、EMTにはERK経路とCtBPの両方が関与しますが、両者がどのようにして協調的に作用し、EMTを誘導するのか、その分子メカニズムに関してはこれまで全く知見がありませんでした。
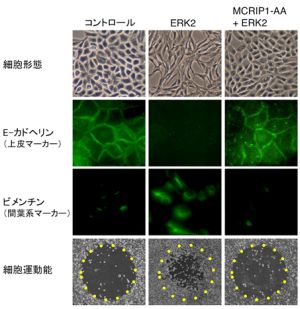
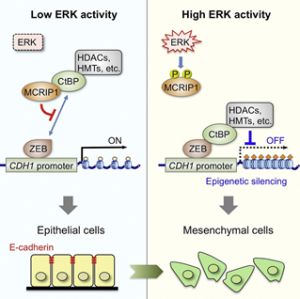 今回私たちは、ERKによってリン酸化される基質分子を、ヒトcDNAライブラリーから網羅的に探索する新たな遺伝子スクリーニング法(酵母3-hybrid法)を開発して、これまで全く報告のない新規ERK基質分子(MAPK-regulated co-repressor interacting protein 1: MCRIP1と命名)を同定することに成功しました。さらに、MCRIP1の生理機能の解析を行い、MCRIP1がCtBPと直接結合すること、またその結果、CtBP(転写抑制共役因子)とZEB(転写抑制因子)間の結合を競合的に阻害して、CtBP依存的な転写抑制を解除する作用を持つことを見出しました(図1左)。さらに興味深いことに、ERKによってMCRIP1がリン酸化されると、MCRIP1がCtBPから解離して、CtBPがZEBと結合できる様になり、最終的にE-カドヘリンの発現抑制とEMTが誘導されることを明らかにしました(図1右)。また、ERKでリン酸化されないMCRIP1点変異体(MCRIP1-AA)を細胞に発現させると、増殖因子刺激を与えてもMCRIP1がCtBPから解離しないため、E-カドヘリンの発現が低下せずEMTも起こらなくなることを確認しました(図2)。以上の結果から、ERKがMCRIP1のリン酸化を介してCtBPの転写抑制作用を調節することで、E-カドヘリンの発現とEMTが制御されていることが明らかとなりました。
今回私たちは、ERKによってリン酸化される基質分子を、ヒトcDNAライブラリーから網羅的に探索する新たな遺伝子スクリーニング法(酵母3-hybrid法)を開発して、これまで全く報告のない新規ERK基質分子(MAPK-regulated co-repressor interacting protein 1: MCRIP1と命名)を同定することに成功しました。さらに、MCRIP1の生理機能の解析を行い、MCRIP1がCtBPと直接結合すること、またその結果、CtBP(転写抑制共役因子)とZEB(転写抑制因子)間の結合を競合的に阻害して、CtBP依存的な転写抑制を解除する作用を持つことを見出しました(図1左)。さらに興味深いことに、ERKによってMCRIP1がリン酸化されると、MCRIP1がCtBPから解離して、CtBPがZEBと結合できる様になり、最終的にE-カドヘリンの発現抑制とEMTが誘導されることを明らかにしました(図1右)。また、ERKでリン酸化されないMCRIP1点変異体(MCRIP1-AA)を細胞に発現させると、増殖因子刺激を与えてもMCRIP1がCtBPから解離しないため、E-カドヘリンの発現が低下せずEMTも起こらなくなることを確認しました(図2)。以上の結果から、ERKがMCRIP1のリン酸化を介してCtBPの転写抑制作用を調節することで、E-カドヘリンの発現とEMTが制御されていることが明らかとなりました。
 さらに私達は、ヒト癌におけるMCRIP1のリン酸化状態を解析したところ、B-Rafなどの癌遺伝子の作用により、ERK経路が恒常的に活性化している多くの癌細胞で、MCRIP1が異常にリン酸化されていること、またその結果MCRIP1がCtBPと結合する能力を失ってEMTが起こり易い状態(即ち、癌細胞の浸潤・転移が起きやすい状態)になっていることを見出しました。
さらに私達は、ヒト癌におけるMCRIP1のリン酸化状態を解析したところ、B-Rafなどの癌遺伝子の作用により、ERK経路が恒常的に活性化している多くの癌細胞で、MCRIP1が異常にリン酸化されていること、またその結果MCRIP1がCtBPと結合する能力を失ってEMTが起こり易い状態(即ち、癌細胞の浸潤・転移が起きやすい状態)になっていることを見出しました。
これまで、ERK経路が発癌を導くメカニズムとして、主に、ERKが細胞増殖に重要な遺伝子群(fos/jun等の初期応答遺伝子や、サイクリンD等)の発現を亢進させる作用を持つことが注目されてきました。しかしながら近年、ERKは、増殖を促進する遺伝子の発現を亢進させるだけでなく、同時に、増殖阻害分子や癌抑制遺伝子の発現をも抑制して、癌の更なる悪性化を導くことが明らかになってきました。しかしながら、ERKシグナルがどの様にして特定の遺伝子の発現を抑制するのか、その分子機構に関しては殆ど知見がありませんでした。本研究により、新たなERK基質分子MCRIP1が同定されると共に、ERKがMCRIP1を介して転写抑制共役因子CtBPの機能を制御することでヒストン修飾を変化させ、癌抑制遺伝子E-カドヘリンのジーン・サイレンシングが惹起されることが明らかとなりました。また、癌で認められるMCRIP1のリン酸化異常が、癌細胞のEMTを促進し、癌の浸潤・転移を亢進させる一因となることが明らかになりました。今後、今回の研究成果を応用した新たな癌治療薬の開発が期待されます。
図1:ERKおよびCtBPによるEMT制御機構
1) 無刺激状態でERK活性が低い場合、MCRIP1がCtBPと結合することで、CtBP-ZEB分子間の会合を競合的に阻害し、CtBPがプロモーター上にリクルートされるのを防いでいる。この結果、E-カドヘリンの発現が保持され、上皮細胞としての性質が保たれる。2) 一方、増殖因子によってERKが活性されると、MCRIP1がリン酸化されてCtBPから解離する。その結果、フリーとなったCtBPはZEBと結合して、HDACやHMTと共にプロモーター上にリクルートされ、ヒストン修飾を変化させてE-カドヘリンの発現を抑制し、EMTを誘導する。
図2:MCRIP1-AA変異体によるERK依存的上皮間葉転換の阻害