
| 発表のポイント |
|---|
|
概要
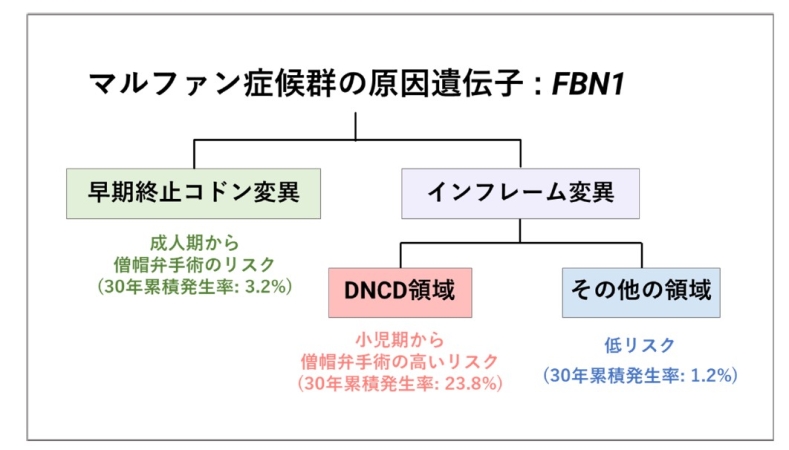
東京大学医学部附属病院の小児科およびマルファン症候群センターの研究グループは、マルファン症候群(注1)の患者において、遺伝子変異のタイプによって僧帽弁(注2)の手術が必要となるリスクと時期が大きく異なることを明らかにしました。マルファン症候群は約5,000人に1人が罹患する遺伝性疾患で、心臓の弁や大動脈に異常をきたし、手術が必要となることがあります。本研究では437名の患者データを解析し、FBN1遺伝子(注3)の特定領域(DNCD領域、注4)に変異がある患者では、30年間の僧帽弁手術累積発生率が23.8%と、他の変異タイプ(その他の領域の変異1.2%、早期終止コドン変異(注5)3.2%)と比べて著しく高いことを発見しました。また、小児期・思春期から手術リスクが高まる一方、他のタイプでは30歳前後からリスクが上昇するという、年齢依存的な違いも判明しました。
これらの成果により、診断時に行う遺伝学的検査の結果から、個々の患者の将来的な手術リスクと時期を予測することが可能となり、患者・家族への適切な情報提供と、リスクに応じた経過観察計画の立案に役立つことが期待されます。
発表内容
【研究の背景】マルファン症候群は、全身の結合組織に異常をきたす遺伝性疾患で、約5,000人に1人が罹患します。FBN1という遺伝子の異常によって引き起こされ、心臓の弁や大動脈、骨格、眼など多臓器に症状が現れます。中でも僧帽弁の異常は30~65%の患者で認められ、重症化すると16%の患者で手術が必要となる重要な合併症です。
現在、マルファン症候群の診断時には遺伝学的検査が広く行われていますが、遺伝子変異の情報を使って「どの患者が、いつ頃、僧帽弁手術が必要になるのか」を予測することはできませんでした。大動脈疾患については遺伝子変異のタイプによってリスクが異なることが知られていますが、僧帽弁疾患については十分に解明されていませんでした。
【研究の内容】
本研究グループは、東京大学医学部附属病院のマルファン症候群センターで2006年から2024年までに診療した437名の患者を対象に、遺伝子変異のタイプと僧帽弁疾患の関係を詳しく調べました。
まず、患者を遺伝子変異のタイプによって3つのグループに分類しました。
1. DNCD領域(エクソン26-37、44-50)に変異があるインフレーム変異(IFV、注6)群
2. その他の領域に変異があるインフレーム変異(IFV)群
3. 早期終止コドン変異(PTC)群
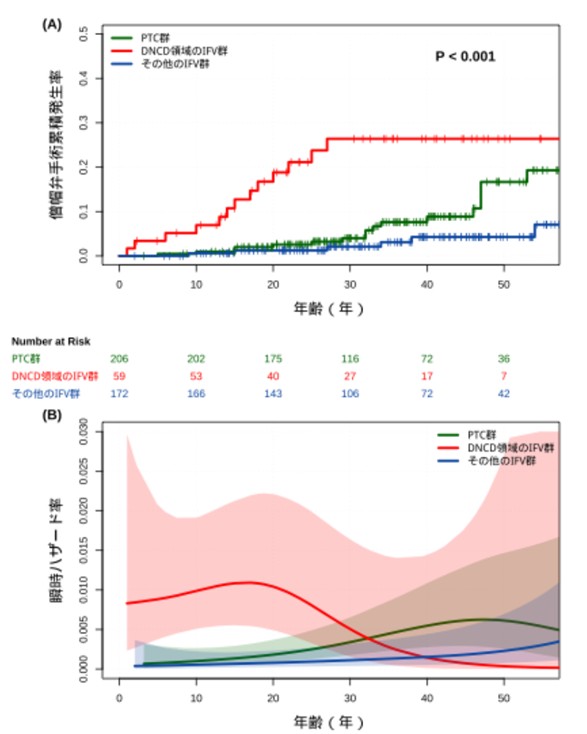
これら3つのグループについて、僧帽弁手術の累積発生率と瞬時ハザード率を比較した結果を図1に示します。主な発見は次のとおりです。
・DNCD領域に変異を持つIFV群では、30年間で23.8%が僧帽弁手術を必要としました。これは、その他の領域の変異(1.2%)や早期終止コドン変異(3.2%)と比べて著しく高い割合です。
・DNCD領域変異患者では、小児期・思春期から手術リスクが高まることが判明しました。
・一方、PTC群では、30歳前後から遅れてリスクが上昇する傾向が見られました。
・30歳以下の患者において、DNCD領域変異は他のタイプと比較して約8倍高い手術リスクを示しました。
【研究の意義】
本研究成果により、以下のような臨床的な意義が期待されます。
1. 個別化医療の実現:診断時の遺伝学的検査結果から、患者ごとの僧帽弁手術リスクを予測できます。
2. 適切な経過観察計画:リスクの高いDNCD領域変異患者には小児期から頻回の観察を、リスクの低い患者にはより緩やかな観察を提案できます。
3. 患者・家族への情報提供:より正確な予後情報を提供することで、人生設計や治療選択の意思決定を支援できます。
なお、本研究は東京大学大学院医学系研究科・医学部倫理委員会の承認のもと実施されました。
発表者・研究者等情報
東京大学
大学院医学系研究科 生殖・発達・加齢医学専攻 小児医学講座
犬塚 亮 准教授
兼:東京大学医学部附属病院 小児科
兼:東京大学医学部附属病院 マルファン症候群センター
医学部附属病院 小児科
河島 裕樹 助教
医科学研究所附属病院 内科系診療部門 循環器内科
武田 憲文 講師
兼:同研究所附属病院 内科系診療部門 循環器内科 科長
兼:東京大学医学部附属病院 マルファン症候群センター センター長
大学院医学系研究科 生殖・発達・加齢医学専攻 小児医学講座
犬塚 亮 准教授
兼:東京大学医学部附属病院 小児科
兼:東京大学医学部附属病院 マルファン症候群センター
医学部附属病院 小児科
河島 裕樹 助教
医科学研究所附属病院 内科系診療部門 循環器内科
武田 憲文 講師
兼:同研究所附属病院 内科系診療部門 循環器内科 科長
兼:東京大学医学部附属病院 マルファン症候群センター センター長
論文情報
雑誌名:Journal of the American College of Cardiology(JACC)題 名:Genotype-Guided Risk Stratification of Mitral Valve Surgery in Marfan Syndrome
著者名:Yuki Kawashima, Norifumi Takeda, Akiharu Omori, Yoshitsugu Nogimori, Kazuhiro Shiraga, Hitomi Masuda, Hiroki Yagi, Yuki Taniguchi, Haruo Yamauchi, Minoru Ono, Ryo Inuzuka*
(*責任著者)
DOI:10.1016/j.jacc.2026.01.059
URL:https://www.doi.org/10.1016/j.jacc.2026.01.059
用語解説
(注1)マルファン症候群FBN1遺伝子の異常により引き起こされる遺伝性疾患。約5,000人に1人が罹患し、心臓・血管・骨格・眼など全身の結合組織に異常をきたします。心臓では大動脈瘤・大動脈解離や僧帽弁逆流などが生じ、重症例ではいずれも手術が必要となります。
(注2)僧帽弁
心臓の左心房と左心室の間にある弁。血液の逆流を防ぐ役割を持ちます。マルファン症候群では僧帽弁が粘液状に変性し、逆流(僧帽弁閉鎖不全症)を起こすことがあります。
(注3)FBN1遺伝子
フィブリリン-1というタンパク質を作る遺伝子。フィブリリン-1は結合組織の主要な構成成分で、組織に弾力性と強度を与えます。この遺伝子に変異があると、組織の構造が弱くなり、マルファン症候群のさまざまな症状が現れます。
(注4)DNCD
Dominant Negative variants affecting Cysteine residues and in-frame Deletionsの略。FBN1遺伝子のエクソン26-37および44-50の領域で、システイン残基に影響する変異やフレーム内欠失を指します。この領域はカルシウム結合上皮成長因子様ドメインを含み、フィブリリン-1の構造維持に重要な役割を果たしています。
(注5)早期終止コドン変異(PTC:Premature Termination Codon)
遺伝子の変異により、タンパク質の合成が途中で止まってしまい(ナンセンス変異)、通常より短いタンパク質ができるタイプの変異です。多くの場合、この短いタンパク質は不安定で分解されやすく、結果として正常なタンパク質の量が減少する「ハプロ不全」という状態になります。大動脈疾患のリスクが高いことが知られています。
(注6)インフレーム変異(IFV:In-Frame Variant)
遺伝子の変異により、タンパク質を構成するアミノ酸の一部が別のアミノ酸に置き換わったり(ミスセンス変異)、一部のアミノ酸が欠けたり(フレーム内欠失)するタイプの変異。タンパク質の長さは正常と同じですが、構造が異常になります。異常な構造を持つタンパク質が正常なタンパク質の働きを邪魔する「ドミナントネガティブ効果」により、症状が出ると考えられています。
問合せ先
<研究内容について>
東京大学大学院医学系研究科 生殖・発達・加齢医学専攻 小児医学講座
(東京大学医学部附属病院 小児科)
准教授 犬塚 亮(いぬづか りょう)
<機関窓口>
東京大学医学部附属病院 パブリック・リレーションセンター
東京大学医科学研究所 プロジェクトコーディネーター室(広報)