| Points |
|---|
|
Overview
Collaborating teams of researchers from Tohoku Medical Megabank Organization (ToMMo)※1 at Tohoku University, Iwate Tohoku Medical Megabank Organization (IMM)※2 at Iwate Medical University, RIKEN, and the Institute of Medical Science, the University of Tokyo (BBJ) ※3 carried out the variant detection on Whole Genome Sequence (WGS) Data, which was collected by ToMMo/IMM and BBJ (4,495 and 3,114 sequences respectively, total 7,609).Those analyses have published as the GEM Japan Whole Genome Aggregation (GEM-J WGA) panel, including variant frequencies for 7,609 people as unrestricted-access data, through TogoVar※4, a database developed by the National Bioscience Database Center (NBDC), the Japan Science and Technology Agency (JST).
The results of individual genome sequences mapped to reference genome sequences※5 will also be published soon as controlled-access/group-sharing data, through the Japanese Genotype-phenotype Archive (JGA) / AMED Genome group sharing Database (AGD)※6 of the Bioinformation and DDBJ Center at the National Institute of Genetics.
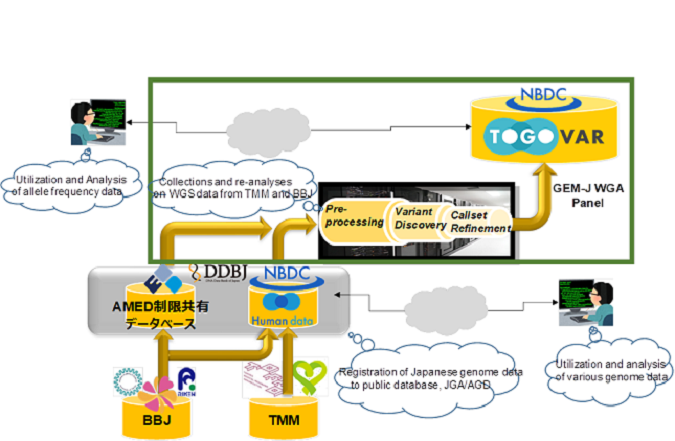
Figure 1. Workflow for GEM-J WGA panel via re-analyzing WGS data of TMM and BBJ
Background
Currently, as next-generation sequencers drastically reduce the cost of genome sequencing, genomic research for disease identification and pathology is advancing through the discovery of several disease-related variants in DNA sequence data obtained from patient specimens (blood, tissue samples, etc.). Based on these accumulating results, genomic medicine has already been realized at the clinical level in the field of cancer.Although millions of variants are detected in WGS data obtained from the specimens provided from participants in genomic medical research, variants that are causative of disease and those of unknown significance for disease (VUS: Variant of Uncertain Significance) are mixed. As those variants exist in the general population at a certain frequency, for the purposes of personalized medicine for monogenic diseases it is important to consider a “frequency filter” to exclude variants that are unlikely to have a significant health impact. This frequency filter requires exact variant allele frequencies in the general population.
As it has been reported that WGS produces less biased results than whole exome sequences in the translational region of the genome※7, it would be desirable to maintain a variant allele frequency database obtained by WGS, so that many researchers can use the data for future clinical applications. Mutation, genetic drift, migration, and natural selection lead to changes in allele frequencies of variants, reflecting the genetic diversity of the biological population.
However, allele frequencies for extremely rare variants fluctuate within the biological population. Therefore, it would be desirable to use allele frequency data derived from a population with the same genetic background in developing the frequency filter. Furthermore, as genetic drift and migration greatly affect variant allele frequencies in different regions of Japan, it is an important for the realization of Japanese genomic medicine to acquire data that reflect the general population across all areas of Japan.
Allele frequency data is also used to compare allele frequencies in other populations when studying disease-causing variants. However, as various tools have been developed and utilized for variant detection, it is also important to adopt international standard methods and thereby construct comparable data sets.
Achievements
To construct the variant frequency panel, sequence data were integrated and analyzed from: 4,307 research volunteers from a cohort study of the general population of Miyagi and Iwate prefectures conducted by the TMM project, 2,857 patient volunteers in hospitals collaborating with the Tailor-Made Medical Treatment Programs※8 conducted by RIKEN and the Institute of Medical Science, the University of Tokyo (Biobank Japan, BBJ), 188 research volunteers at the National Hospital Organization Nagasaki Medical Center, and 257 research volunteers from the RIKEN Center for Integrative Medical Sciences (Table 1). In order to create internationally comparable data based on WGS of these research volunteers, those analyzed data-sets were mapped to the GRCh37※5 reference genome sequence, and variant detection was carried out using the GATK(Genome Analysis Toolkit)※9 standards.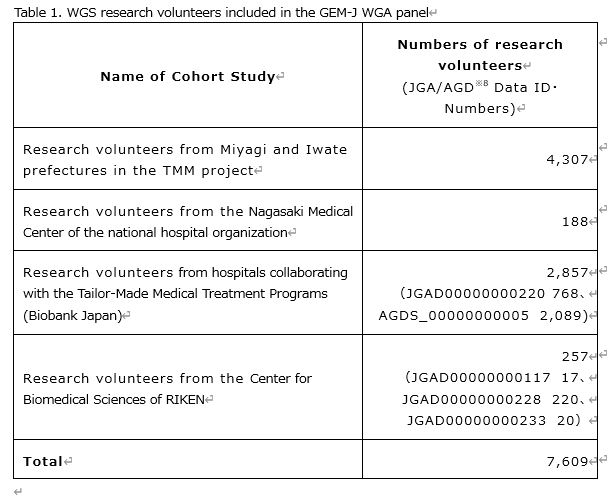
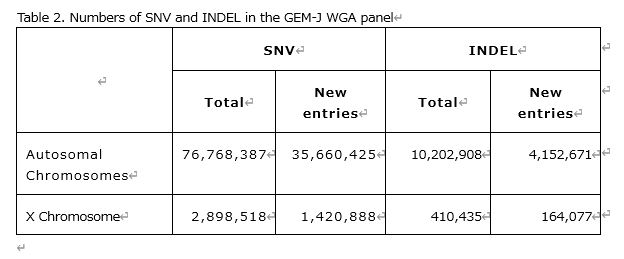
As a result, 76,768,387 single nucleotide variations (Single Nucleotide Variation: SNV)and 10,202,908 insertion and deletion sequences (Insertion and Deletion:INDEL) on autosomal chromosomes were detected. On the X chromosome, 2,898,518 SNVs and 410,435 INDELs were detected (Table 2).
In addition, based on the obtained variant data for each individual, we conducted a principal component analysis referring to the 1,000 Genome Project※10 to confirm the genetic background of the Japanese population. Furthermore, quality control was carried out by comparing genetic distances between individuals and eliminating relatives that biased variant frequency information.
This project is an initiative of the GEnome Medical alliance Japan (GEM Japan, GEM-J)※11, which is a driver project of The Global Alliance for Genomics and Health (GA4GH) ※12, an international coalition that promotes international data sharing of genomic and clinical information. It is expected that the publication of the variant frequency panel for the Japanese population will contribute to international research on intractable and rare diseases.
Since analysis of variant data started with the international 1,000 Genome Project, various types of population/aggregation data have been analyzed, including through gnomAD※13 etc. In Japan, where the population forms a distinct ethnic group, data from more than ten thousand people will be necessary to perform high accuracy diagnosis and to develop genomic medicine.
In international genome research, many European genome data sets have been published, whereas there are very few East Asian data sets. To perform genomic diagnostics for East Asian populations, or for “Filtered” genomic diagnostics in multiple ethnic groups, sharing of East Asian data sets, including those from Japan, is much in demand.
Future Outlook
The results of this project are expected to serve as basic data for future genomic medicine initiatives and to be useful for the following types of research:- To improve the accuracy of identification of the causative variants for intractable and rare diseases, by improving the accuracy of frequency filters.
- To improve the accuracy of imputation of rare variants based on rare haplotype information via a larger reference panel※14.
Glossary and References
※1 Tohoku Medical Megabank Organization (ToMMo):https://www.amed.go.jp/en/program/list/14/01/002.html
https://www.megabank.tohoku.ac.jp/english/about/outline/
※2 Iwate Tohoku Medical Megabank Organization (IMM):
http://iwate-megabank.org/en/about/mission
※3 Biobank Japan (BBJ):
https://biobankjp.org/english/pdf/english.pdf
※4 TogoVar:
https://togovar.biosciencedbc.jp/doc/about?locale=en
※5 Reference genome sequences (derived from The Genome Reference Consortium) :
https://www.ncbi.nlm.nih.gov/grc
※6 Japanese Genotype-phenotype Archive (JGA) / AMED Genome group sharing Database (AGD):
https://www.ddbj.nig.ac.jp/jga/index-e.html
https://www.ddbj.nig.ac.jp/agd/index-e.html
https://humandbs.biosciencedbc.jp/en/
※7 Lelieveld S. H. L. et al, “Comparison of Exome and Genome Sequencing Technologies for the Complete Capture of Protein-Coding Regions” Hum Mutat. 2015; 36 (8): 815-822. doi: 10.1002/humu.22813.
https://www.ncbi.nlm.nih.gov/pubmed/25973577
※8 The Tailor-Made Medical Treatment Programs:
https://www.amed.go.jp/en/program/list/04/01/003.html
※9 GATK(Genome Analysis Toolkit):
https://github.com/gpc-gr/panel3552-scripts
※10 The 1,000 Genomes :
https://www.internationalgenome.org/about
※11 GEnome Medical alliance Japan (GEM Japan, GEM-J):
https://www.amed.go.jp/en/aboutus/collaboration/ga4gh_gem_japan.html
※12 The Global Alliance for Genomics and Health (GA4GH):
https://www.ga4gh.org/
※13 gnomAD(The Genome Aggregation Database):
https://gnomad.broadinstitute.org/about
※14 McCarthy S. et al., “A reference panel of 64,976 haplotypes for genotype imputation” Nat Genet 2016, 48 (10):1279-1283. doi: 10.1038/ng.3643.
https://pubmed.ncbi.nlm.nih.gov/27548312/
