研究内容
当研究室では、より効果的なワクチン開発に役立てることを目的として、生体によるウイルス認識機構、インフルエンザウイルス感染モデルを用いた粘膜免疫制御機構と腸内細菌によるウイルス特異的免疫応答の制御機構について研究を行っております。
1.自然免疫系によるウイルス認識機構
⇒生体が目には見えないウイルスをどう異物として認識しているのか?
・Moriyama et al. iScience. 2020 [Link]
・Moriyama et al. Nat Commun. 2019 [Link] [プレスリリース]
・Chen et al. Front Microbiol. 2019 [Link]
・Ichinohe et al. PNAS. 2013 [Link] [プレスリリース]
・Ito et al. PLoS Pathog. 2012 [Link]
・Ichinohe et al. Nat Immunol. 2010 [Link]
2.腸内細菌叢によるウイルス性肺炎の重症化抑制機構
⇒腸内細菌叢がウイルス性肺炎の重症化の抑制にどう役立っているのか?
・Nagai et al. Nat Commun. 2023 [Link] [プレスリリース]
・Moriyama et al. PNAS. 2019 [Link] [プレスリリース]
・Ichinohe et al. PNAS. 2011 [Link]
3.ウイルスの宿主免疫回避機構
⇒ウイルスは宿主の免疫をどう回避しているのか?
・Moriyama et al. J Virol. 2018 [Link]
・Moriyama et al. J Virol. 2016 [Link]
・Komune a et al. J Virol. 2011 [Link]
4.ウイルスの病原性発現機構
⇒インフルエンザウイルスや新型コロナウイルスがヒトや動物を重症化させるメカニズムは何か?
⇒武漢株、デルタ株、オミクロン株で重症度に違いがあるのはなぜか?
・Kobayashi et al. PLoS Pathog. 2024 [Link]
5.ウイルス感染による重症化メカニズムの解明
⇒なぜ高齢者ではインフルエンザウイルスや新型コロナウイルスに感染すると重症化しやすいのか?
・Nagai et al. Nat Commun. 2023 [Link] [プレスリリース]
6.ウイルスと宿主の共生メカニズム
⇒水きん類(アヒルやカモなどの水鳥)はなぜインフルエンザウイルスと共生できるのか?
⇒新型コロナウイルスに感染しても無症状のひとがいるのはなぜか?
・Nagai et al. Nat Commun. 2023 [Link] [プレスリリース]
7.インフルエンザウイルスや新型コロナウイルスに対する経鼻ワクチンの開発
⇒鼻に噴霧してウイルスの感染そのものを阻止する次世代ワクチンの研究
・Kobayashi et al. NPJ Vaccines. 2025 [Link]
・Nagai et al. mBio. 2021 [Link] [プレスリリース]
・Ichinohe et al. PNAS. 22011 [Link]
・Ichinohe et al. J Exp Med. 2009 [Link]
8.±1℃の生体防御学
⇒発熱の意義とは何か?1℃の違いがウイルス増殖に与える影響は?
・Nagai et al. Nat Commun. 2023 [Link] [プレスリリース]
・Moriyama et al. PNAS. 2019 [Link] [プレスリリース]
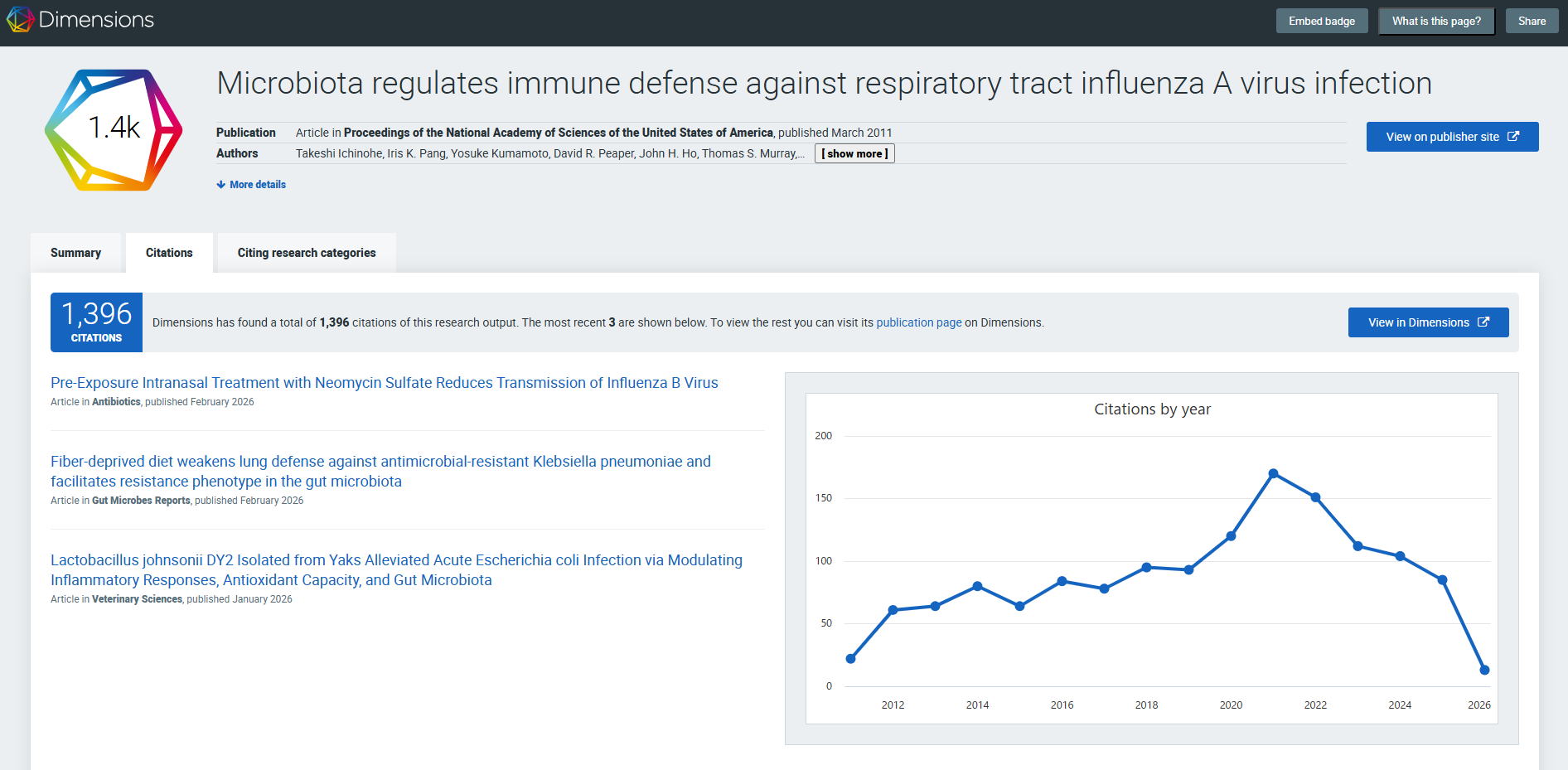
参考:論文引用回数(Ichinohe et al. PNAS 2011)