主な研究成果(2006年以降)
1:
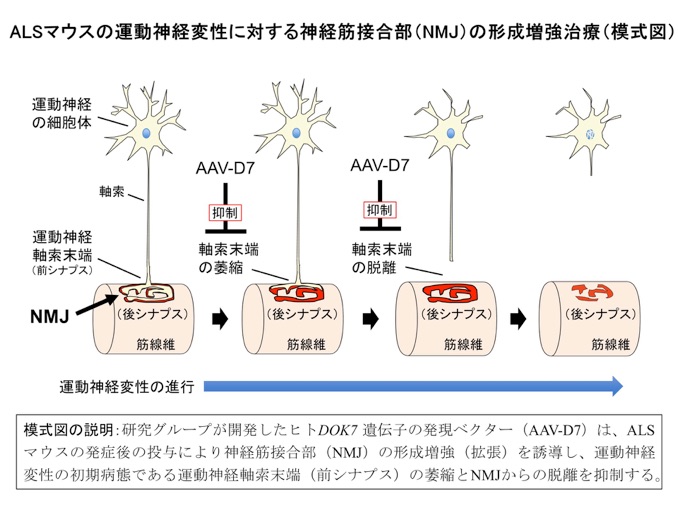
我々の運動機能には、運動神経を介した骨格筋収縮の緻密な制御が必要であり、両者は神経筋接合部(NMJ:Neuromuscular Junction)と呼ばれるシナプス(神経筋シナプス)により結ばれている(模式図、左端)。当然のことながら、NMJの喪失は呼吸を含めた運動機能の喪失を意味する。我々は、このように重要なNMJの形成に不可欠のタンパク質としてDok-7を発見し(参考文献:英文原著10/和文6−9)、その劣性遺伝病としてNMJの形成不全病であるDOK7型筋無力症を発見した(参考文献:英文原著9)。さらに、DOK7発現ベクターの投与によるNMJ形成の増強(NMJの拡張)が、NMJ形成不全を呈するDOK7型筋無力症や常染色体優性エメリードレフュス型筋ジストロフィー(AD-EDMD)のモデルマウスに有効な治療戦略(NMJ形成増強治療)になることを実証した(参考文献:英文原著4)。近年、このようなNMJ形成不全との関連が、上記のDOK7型筋無力症やAD-EDMD等の筋原性疾患(ミオパチー)だけでなく、筋萎縮性側索硬化症(ALS)などの神経原性疾患(ニューロパチー)や加齢性の筋萎縮などにおいても報告されているが、その治療標的としての可能性については不明な部分が多い。
以上の経緯を踏まえ、我々は現在に至るまで根本的な治療法が見出されていない運動神経変性疾患であるALSのモデルマウスを対象に、NMJ形成増強治療の有効性を検討した。具体的には、運動機能の低下を示したALSモデルマウスにDOK7発現ベクターを投与することで、1)NMJ形成不全(模式図に記載の運動神経軸索末端の萎縮とNMJからの脱離)の抑制、2)筋萎縮の抑制、3)運動機能の改善、4)生存期間の延長、を実証した(参考文献:英文原著1)。これらの成果は、独自に開発したNMJ形成増強治療が、根本的な治療法のないALSに対する新たな治療概念となり得ることを示す発見と言える。さらに、本研究の成果は、NMJ形成不全との関連が解き明かされつつある他の運動神経変性疾患や加齢性の筋萎縮に対する治療法の開発にもつながることが期待される。また、本研究はDOK7発現ベクターを用いた遺伝子治療だけでなく、化合物を用いたNMJ形成増強治療にも道を拓くべき「治療概念の実証研究」としての側面を有している。それ故、老齢マウスを含む様々な疾患モデル動物に対する有効性を検証すると共に、NMJ形成増強効果をもつ化合物の開発、さらには運動神経保護薬などのNMJ以外を標的とする他の薬剤との併用治療に関する研究の推進が求められる。
2:
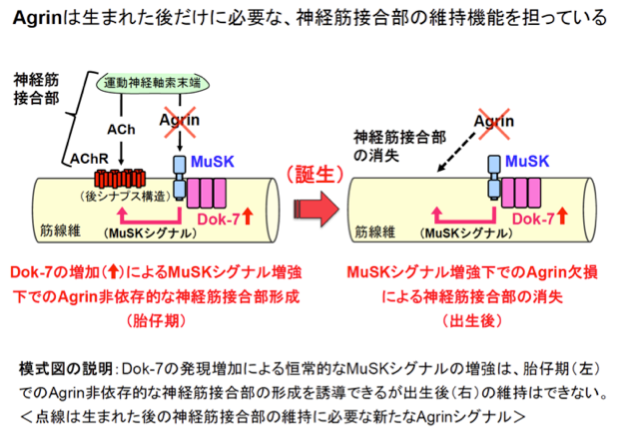
我々の運動機能には、運動神経を介した骨格筋収縮の緻密な制御が必要であり、両者は神経筋接合部(NMJ:Neuromuscular Junction)と呼ばれるシナプス(神経筋シナプス)により結ばれている。当然のことながら、NMJの喪失は呼吸を含めた運動機能の喪失を意味する。我々は、このように重要なNMJの形成に不可欠のタンパク質としてDok-7を発見し(参考文献:英文原著10/和文総説6−9)、その劣性遺伝病としてNMJの形成不全病であるDOK7型筋無力症を発見した(参考文献:英文原著9)。さらに、最近、神経筋接合部の形成不全を伴う様々な疾患に対する治療法として、DOK7発現ベクターの投与による「神経筋接合部の形成増強治療」をマウスにおいて創出した(参考文献:英文原著4)。
他方、我々はDok-7がMuSKというタンパク質(リン酸化酵素)を活性化することで神経筋接合部を形成することも解明しているが(参考文献:英文原著8/和文総説3−5)、MuSKにはAgrinと言う別の活性化因子の存在が古くから知られており、両者の違いは未解明であった。そこで、今回、Dok-7を多量に産生することでMuSKの活性化を恒常的に増強するマウスを作出し、そのマウスにAgrin欠損を導入したところ、胎仔期に形成された神経筋接合部が誕生後の数週間で消失してしまうことが判明した(模式図)。この事実は、胎仔期のAgrin機能がDok-7の発現増強によるMuSKの活性化で補えるのに対して、生まれた後のAgrinはMuSK活性化以外の、Dok-7では代替不可能な仕組みで神経筋接合部を守っていることを意味する(参考文献:英文原著3)。以上の成果は、我々が母体内で形成した神経筋接合部をどのようにして維持しているのかを理解するための鍵となる知見であり、また、その多くが生まれた後に発症する多様な神経・筋疾患の理解や制御につながる発見と言える。
3:
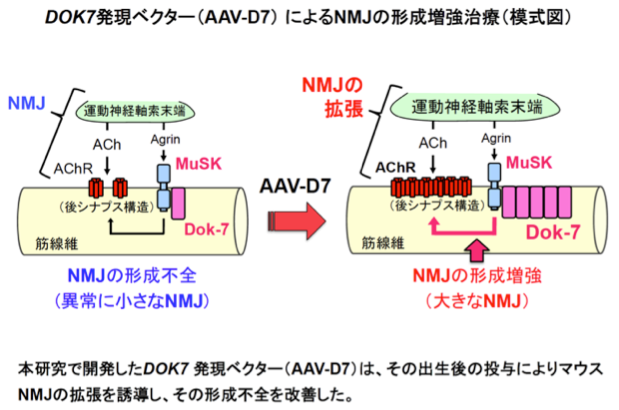
我々の運動機能には、運動神経を介した骨格筋収縮の緻密な制御が必要であり、両者は神経筋接合部(NMJ:Neuromuscular Junction)と呼ばれるシナプス(神経筋シナプス)により結ばれている。当然のことながら、NMJの喪失は呼吸を含めた運動機能の喪失を意味する。我々は、このように重要なNMJの形成に不可欠のタンパク質としてDok-7を発見し(参考文献:英文原著10/和文総説6−9)、その劣性遺伝病としてNMJの形成不全病であるDOK7型筋無力症を発見した(参考文献:英文原著9)。そこで、NMJの形成不全を伴う神経筋疾患に関する情報を広く検討したところ、NMJ疾患として知られる筋無力症の枠を超え、筋ジストロフィー、筋萎縮性側索硬化症(ALS)、脊髄性筋萎縮症(SMA)や加齢性筋肉減少症(Sarcopenia)などの多様な神経筋疾患にもNMJの形成不全が認められることに気づいた。これらの知見を踏まえ、我々はDok-7タンパク質の人為的な発現増強による「NMJ形成増強治療の創出」、つまり「小さくなってしまったNMJを大きくする治療の創出」を進めた(参考文献:英文原著4/和文総説2)。Dok-7の発現増強の手法としては、ヒト骨格筋での安全、かつ、長期にわたる外来遺伝子発現に優れたアデノ随伴ウイルス(AAV: adeno- associated virus)を選び、Dok-7発現ベクター(AAV-D7)を作成した。その上で、AAV-D7をDOK7型筋無力症モデルマウスの発症後に投与したところ、形成不全を呈するNMJの拡張、運動機能の改善、早期致死性の回避をもたらした。さらに、DOK7遺伝子そのものではなく、核内分子をコードするLMNA遺伝子の異常によるエメリードレフュス型筋ジストロフィー(AD-EDMD: autosomal dominant Emery-Dreifuss muscular dystrophy)のモデルマウスに対しても、AAV-D7の発症後の投与はNMJの拡張、運動機能の改善、延命効果をもたらした。以上の成果は、1)哺乳動物のNMJを後天的、人為的、かつ安全に拡張する方法の創出と、2)NMJの形成不全を伴う多様な神経筋疾患に対する新規治療概念の確立を意味するものである。
4:
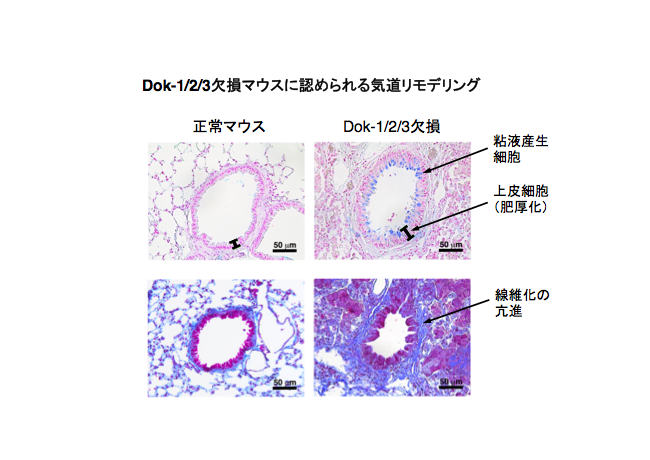
我々の生命活動に必須の呼吸機能は、骨格筋の収縮・弛緩が生み出す適切な気流の維持によって支えられている。それ故に、仮に骨格筋機能が正常であったとしても、気道構造の変化(リモデリング)による気流の障害は重篤な疾病の原因となる。喘息は肺組織の慢性的な炎症による気流障害を伴う呼吸器疾患であり、炎症性サイトカインの産生を伴う気道リモデリングや気道過敏性を特徴とする。今回、我々はDok-1/2/3欠損マウスが特定のアレルギー刺激の非存在下に気道過敏性の亢進や気道リモデリングを伴う喘息様病態を呈することを明らかにした(参考文献:英文原著6)。例えば、Dok-1/2/3欠損マウスの気道(細気管支)ではアルシアンブルーにより染色(青色)される粘液産生細胞の増多と気道上皮の肥厚が示されている(図の上段、矢印)。また、マッソントリクローム染色により染色(青色)される線維化の亢進も明らかにされている(下段、矢印)。以上の知見はDok-1/2/3が協調的に気道リモデリングや気道過敏性の亢進による気流障害の抑制に機能していることを示している。
5:
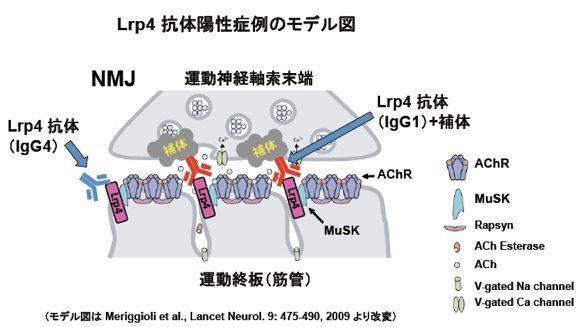
重症筋無力症(Myasthenia Gravis)は異常な自己抗体によって発症する疾患であり、骨格筋収縮に必須のシナプスである神経筋接合部(NMJ)の構造・機能不全による筋力低下と易疲労性を伴う難病である。その多くは、NMJの筋管側に局在するアセチルコリン受容体(AChR)に対する抗体をもつが、同じくNMJの筋管側に局在するMuSKに対する抗体をもつ症例も報告されている。これらの自己抗体は、補体を介したNMJの破壊や標的分子の機能阻害によって筋無力症を惹起すると理解されている。今回、我々は重症筋無力症の新たな病原性自己抗体として抗 Lrp4 抗体を発見し、AChRやMuSK抗体を持たない症例に対する診断・治療法への道を拓いた(参考文献:英文原著7)。上記のモデル図に記載の通り、抗Lrp4抗体の多くは補体の活性化による組織破壊能をもつIgG1であると共に、NMJの形成・維持に必須のLrp4とその活性化因子(Agrin)との結合を阻害する活性をもつ自己抗体である。
6: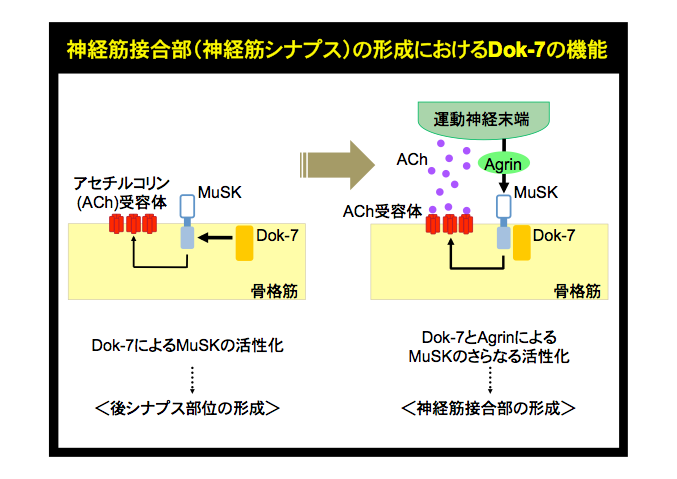
これまでの研究によって、我々は神経筋接合部の形成に必須のタンパク質としてDok-7を同定し、ヒトDOK7遺伝子の異常によって神経筋接合部の形成不全を伴う筋無力症(DOK7型筋無力症)が発症することを明らかにしている(参考文献:英文原著9、10/和文総説6−9)。しかしながら、Dok-7がどの様な動作原理の下で神経筋接合部の形成に機能しているかについては不明な部分が多く、治療法開発の基盤となる情報が求められていた。今回、我々はDok-7が神経筋接合部の形成を司るMuSKと言う受容体型チロシンキナーゼに直接、細胞内から働きかける筋内在の活性化因子であり、Dok-7を失った筋管ではMuSKが活性化されないことを解明した。さらに、このDok-7シグナルを人為的に増強することでマウスにおける神経筋接合部の形成が促進されることも明らかにした。これらの知見は、DOK7型筋無力症やMuSKの機能低下を伴う筋無力症の治療法開発にむけた手がかりとなるべき重要な情報であり、また、受容体型チロシンキナーゼが細胞外からの刺激を細胞内シグナルに変換すると言う原則に従わない、全く新しいシグナル制御機構の発見として、高度の学術的な意義をもつ(参考文献:英文原著8<当該号の表紙は本論文に関する顕微鏡写真です>/英文総説2/和文総説3−5)。事実、最近、受容体型チロシンキナーゼの2例目の細胞内活性化因子としてcytohesinが同定され、その機能阻害が標的PTKであるErbBに依存的な癌細胞の増殖を個体レベルで抑制することが報告されている(Cell, 143: 201-211, 2010)。
7:
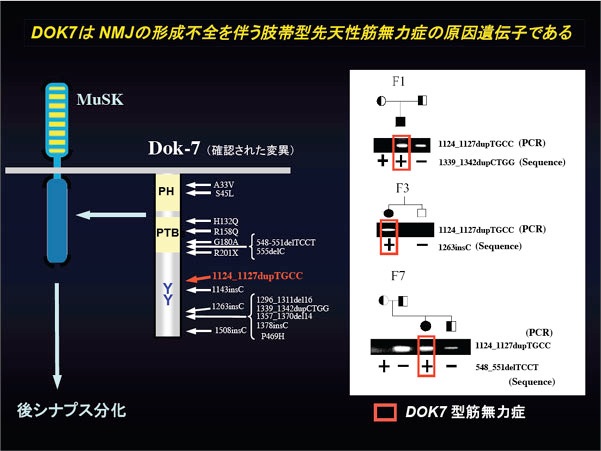
David Beeson教授らの症例についてdok-7遺伝子変異を探索したところ、その両アレル性の異常が神経筋接合部(NMJ)の形成不全を伴う先天性筋無力症候群の発症と一致することが判明した(右パネル)。また、最も多くの症例に認められるC末端欠失変異(1124_1127dupTGCC)がDok-7による後シナプス構造の誘導機能を著しく減弱させることも明らかとなった。これらの知見から、dok-7がNMJ形成不全型CMS(DOK7型筋無力症)の原因遺伝子であると結論された(参考文献:英文原著9/和文総説7−9)。
8:
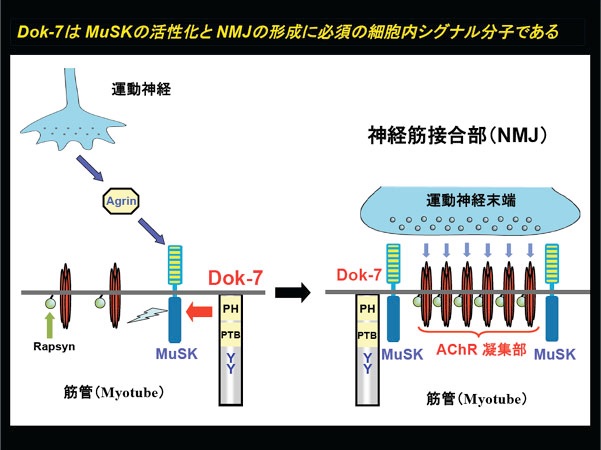
我々がDokファミリー分子の新たなメンバーとして単離したDok-7は、骨格筋収縮の運動神経支配に必須のシナプスである神経筋接合部(NMJ: Neuromuscular Junction)の後シナプス部位に局在し、受容体型チロシンキナーゼであるMuSKの活性化に必須の役割を担っている。MuSKの機能が神経筋接合部の後シナプス部位の形成に不可欠であることを考えると、この研究成果は、運動神経由来のAgrinと筋内在のDok-7によるMuSKの強い活性化がNMJの形成に必須であることを意味している。事実、我々が世界に先駆けて樹立したDok-7欠損マウスは神経筋接合部を欠き、呼吸を含めた運動機能の欠損を呈した(参考文献:英文原著10/和文総説7−9)。
[参考文献]
英文原著(各論文の詳細は主要原著論文リストを御覧下さい)
-
1.EMBO Mol. Med.: Published online e201607298, 2017
-
2.Biochem. Biophys. Res. Commun.: 478, 135-142, 2016
-
3.Proc. Natl. Acad. Sci. USA.: 111, 16556-16561, 2014
-
4.Science: 345, 1505-1508, 2014
-
5.FEBS Lett.: 587, 3749-3754, 2013
-
6.Genes Cells: 18, 56-65, 2013
-
7.Annals of Neurology: 69, 418-422, 2011
-
8.Science Signaling:2, ra7, 2009
-
9.Science: 313, 1975-1978, 2006
-
10.Science: 312, 1802-1805, 2006
-
11.J. Exp. Med.: 201, 333-339, 2005
-
12.J. Exp. Med.: 200, 1681-1687, 2004
-
13.Genes & Development:14, 11-16, 2000
-
14.Cell: 88, 205-211, 1997
-
15.Science: 251, 192-194, 1991
英文総説
-
1.“Molecular signaling and its pathogenic alterations in neuromuscular junction formation and maintenance” in “Protein Modification in Pathogenic Dysregulation of Signaling (Springer)”: pp. 309-325, 2015
-
2.J. Biochem. : 151, 353-359, 2012 [Full Text]
-
3.Immunol. Reviews: 232, 273-285, 2009
和文総説
-
1.CLINICAL CALCIUM 3月号 「神経筋接合部(NMJ)の形成・維持機構とNMJ形成増強治療」: 27, 413-419, 2017
-
2.実験医学3月号「神経筋接合部の形成不全を伴う神経筋疾患に対する新規治療概念の創出」:p603-606, 2015
-
3.実験医学増刊「シグナル伝達研究の最前線2012-’13」:30, 740−745, 2012
-
4.生体の科学4月号「先天性筋無力症の分子基盤」:62, 106-111, 2011
-
5.実験医学「受容体型キナーゼの細胞内からの活性化:Dok-7による神経筋シナプス形成の分子機構」: p1384-1387, 2009
-
6.実験医学増刊「シグナル伝達研究2008-‘09」:26, 103-110, 2008
-
7.Clinical Neuroscience「神経筋接合部の分子構築」:26, 962-963, 2008
-
8.細胞工学6月号「神経筋シナプス形成と筋無力症におけるDok-7/MuSKシグナル」:p674-678, 2007
-
9.実験医学10月号「神経筋シナプスの形成におけるDok-7の役割:細胞内因子による受容体型キナーゼの活性化」:p2517-2520, 2006
-
10.Molecular Medicine増刊「免疫2006」:42, 230-237, 2005
-
11.実験医学増刊「シグナル伝達研究2005-‘06」:23, 39-47, 2005
-
12.実験医学4月号「血球の恒常性とDok-1/2の新たな機能」:p1126-1132, 2005
-
13.キーワードで理解するシグナル伝達イラストマップ:p18-20, p84-94, 2004
